
Technical Data
技术资料
LiP技术
许多外界干扰都能影响蛋白质的构象,诸如热刺激,蛋白-蛋白相互作用,化合物结合,翻译后修饰等。蛋白构象的变化包括局部波动,形成更大的结构域,折叠构象与非折叠构象,或者单体与多体间的转换等。蛋白结构的变化会影响蛋白功能,例如变构酶的结构变化调控酶的活性。在淀粉样变性病以及一些神经退行性疾病中,某些蛋白的结构变化会导致其形成不溶性沉淀,对细胞及组织造成伤害。
X-ray 结晶、核磁共振(NMR)以及各种光谱学技术能够在体外研究简单蛋白体系的构象变化,却不能胜任复杂生物体系。Forster共振能量转移(FRET)以及细胞内的NMR技术能够在体内监测特定蛋白的构象变化,但这两种技术都需要对蛋白进行标记,不适合大规模研究。基于质谱的蛋白质组学通常被用来研究蛋白丰度的变化,翻译后修饰以及蛋白-蛋白相互作用,殊不知该技术也能高通量研究复杂生物体系中的蛋白结构的变化,而实现该目的需要结合由瑞士分子系统生物研究所(ETH)的Paola Picotti团队开发的limited proteolysis (LiP)技术。本文就为您介绍如何结合该技术及SRM、PRM、DDA、DIA等蛋白质组学技术研究蛋白结构的变化。
LiP-MS原理
LiP-MS检测蛋白构象变化的关键在于LiP技术。该技术的核心是让蛋白处于天然构象的条件下,用广谱性的蛋白酶对其进行酶切。当蛋白构象发生变化时(例如与小分子结合),会暴露出不同的广谱性蛋白酶酶切位点,产生不同的蛋白酶切片段。一次酶切后再将蛋白样品变性,用胰酶进行二次酶切。通过质谱检测,比较不同处理条件的样品中,全酶切及半酶切肽段(LiP酶切)的信号强度变化,进而分析蛋白构象变化(图1)。

图1. 不同构象下,全酶切肽段与半酶切肽段信号强度比较
LiP-MS 工作流程
LiP 样品制备(举例细胞样品):
1. 准备不同处理条件下的样品。Condition1, Condition2, Condition3…
2. 非变性条件下提取蛋白。
细胞裂解液可以选择50mM Hepes, 150mM KCl, 1mM MgCl2, 100mM β-glycerophosphate, 50mM NaF, pH 7.3。机械力破碎或者反复冷冻破碎。3000g,4s℃,离心2min,收集上清。
3. 向蛋白样品中加入广谱性的蛋白酶。
例如蛋白酶K(常用),thermolysin (嗜热菌蛋白酶),subtilisin (枯热杆菌蛋白酶)、papain(木瓜蛋白酶)、糜蛋白酶(chymotrypsin)、或者弹性水解酶(elastase)。这些蛋白酶倾向于剪切未折叠的蛋白区域。酶与蛋白样品的比例要低,例如1:100。酶解时间要短,例如室温酶解5min。在广谱性蛋白酶作用下,这些非变形的蛋白样品会被切割成大的蛋白片段(图2中红色箭头为广谱性酶作用位点)。
4. 蛋白变性,终止酶解反应。
向非变性的蛋白样品中,加入盐酸胍酶至终浓度为7.4M,并且加热3min, 使蛋白样变性并且终止广谱性酶的酶解反应。
5. 胰酶酶解。
将上述变性的蛋白样品按常规流程进行还原、烷基化、胰酶酶解,即加入12.5Mm DTT, 37℃,30min,加入40mM IAA, 室温避光反应45min。样品用100mM NH4CO3 将盐酸胍酶稀释至0.5M, 按酶与底物1:100 (w/w) 加入测序级胰酶,37℃,反应2h,之后再次加入等量胰酶,37℃酶解过夜。
6. 酶解终止。向样品中加入甲酸至pH<3,肽段脱盐干燥,质谱检测。
7. 需要注意的是,每个条件下蛋白样品均需要设定一个对照组,该对照组仅按照常规蛋白组样品制备流程进行变性、还原、烷基化和胰酶酶解(步骤5)。该对照组用于后续数据分析的归一化。不同条件下蛋白构象的变化,在两步酶切中,会产生不同酶切形式的肽段,通过比较肽段的不同酶切模式,可以研究蛋白构象的变化(图2)。
Mass Spectrometry 检测:
LiP得到的肽段样品,根据实验目的,可以依赖多种质谱检测手段进行检测,包括大规模筛选模式(DDA,DIA),或者靶向模式(SRM,PRM)。液相梯度按照常规2~3h分离,质谱检测参数按照常规DDA,DIA, SRM 及PRM 检测模式设置。搜库软件及参数也同常规。需要注意的是,搜库参数中,胰酶酶切需要设定为半酶切模式。定量时选择半酶切且没有漏切的unique肽段,差异倍数至少2倍以上,q<0.02。通常经DDA/DIA筛选得到的构象变化的肽段,需要经靶向验证。
1. Label free DDA 定量:可采用spectra counting 模式,也可采用峰面积模式。
2. DIA定量:采用峰面积模式。
3. SRM、PRM:采用子离子的峰面积模式。
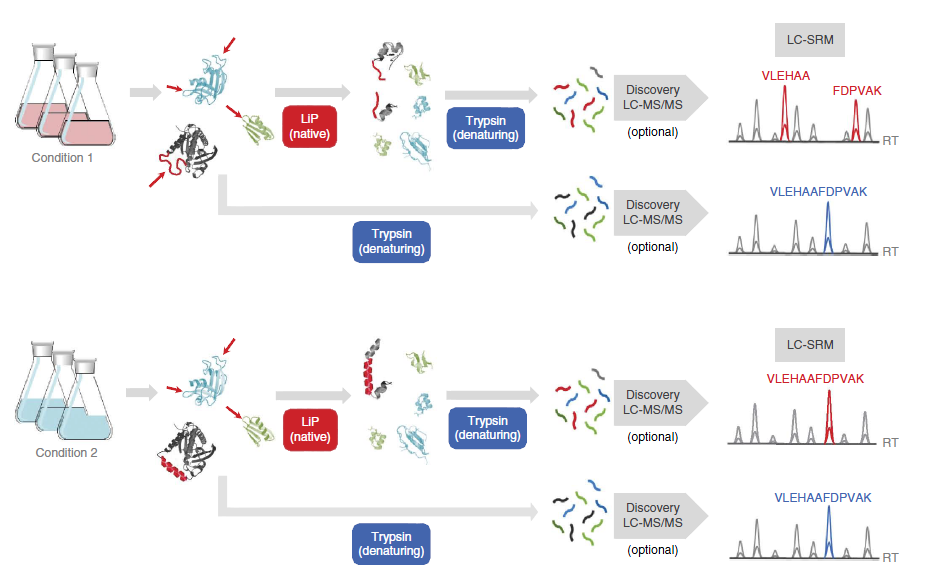
图2. LiP 工作流程
Condition1中的蛋白暴露出蛋白酶K的酶切位点,因此在质谱中可以检测到两个半酶切的肽段。
LiP-MS技术研究蛋白构象的优势
1. 研究体系可以为复杂生物样本,例如细胞、血液、组织。
2. 样本不需要经过富集、提纯及标记。目前在人源细胞中,可检测到约3500个LiP切割位点,包含约1200个蛋白。
3. 可以联合多种质谱检测技术:靶向(PRM,SRM),定性定量(DDA, DIA)。
4. 可以研究特定蛋白的构象变化,也可以高通量研究蛋白网络中的构象变化。
5. 检测灵敏度高,可以检测显著的结构变化,也可以检测微小的结构变化。如果结合更长色谱梯度,可以检测低丰度蛋白的构象变化。
LiP-MS技术的应用
1. 分析翻译后修饰引起的蛋白构象变化;
2. 分析蛋白酶活性的变化(构象变化会导致酶活性的改变);
3. 分析蛋白-蛋白相互作用及网络;
4. 分析蛋白-(代谢、药物)小分子的相互作用及网络;
5. 疾病生物标志物发现(例如神经退行性疾病,淀粉样蛋白/可溶性蛋白的比例)。
LiP-MS技术应用中的注意要点
1. LiP技术必须在非变性条件对蛋白进行酶切,因此只适合与可溶性蛋白,对膜蛋白不适用。
2. 同一体系中,如果某一蛋白存在多种构象,Lip-MS无法区分各个构象。
3. 某些蛋白无论处于何种构象,对LiP剪切都不敏感,因此无法进行构象分析。
Biognosys 拥有LiP的专利技术,在不久的将来,将会推出商业化的产品为科研领域提供技术支持。该部分在正式购买我们的方案后另行培训,如在试用阶段需要了解结果质量,欢迎联系我们:Support@omicsolution.com。
更多内容可参照以下文档:Limited Proteolysis (LiP)–基于蛋白组大规模研究蛋白构象变化
关注我们
服务热线: 18221381405
关注公众号
咨询微信客服

