
Technical Data
技术资料
IPA于近年新增的Analysis Match模块可能很少有人知晓。在当前组学数据越来越多,而IPA的条目摘取主要基于验证性的实验结果,其数据库增长速度远跟不上组学数据的产生速度。Analysis Match模块便是利用来自SRA,GEO,Array express,TCGA等大规模基因组学数据库数据协助IPA分析的新利器,当然由于这些数据的可靠性尚无法和validation实验相提并论,因此暂时还是只能作为一个单独的模块使用。它能够自动发现你提交的IPA Core Analyses是否在生物学结论上和这些数据集相似/相反,从而帮助你通过了解这些研究的背景及其结论协助分析您的实验结果。其方法主要是通过分析结果中的Canonical Pathways, Upstream Regulators, Causal Networks和 Diseases and Functions这些模块结果与数据库中数以千计的实验数据集的Core analysis结果进行相似性比对得到的。
数据来源
Analysis Match约有49000套,来自于SRA, GEO, Array Express, TCGA, LINCS等数据库并进行良好的背景注释的人和小鼠疾病和肿瘤数据集。
结果查看方式
Analysis Match结果以表格形式呈现,并以一个总体相似度分数从高到低排列。
如下图举例

结果表格的最后10列,依次列出了CP(canonical pathway)、UR(upstream regulator)、(Causal Networks)和DE(Downstream effect也就是disease or function)的z-score,4个分析模块综合z-score,以及p-value,4个分析模块综合p-value的结果。
前面的各列则依次列举了相应数据集的来源及实验设计。
在选择感兴趣的数据集(可多选)后,上方工具栏按钮的view as heatmap和view comparison,则可以查看具体各个分析结果的相似性。

背景数据集如何整合入IPA
来自公共数据库的数据集需要经过完整的数据再处理、归一化、质控及注释分析后存入IPA。这些数据可能来自于不同的细胞、组织、检测平台及检测技术。IPA不太可能对这些方面都进行标准化。因此,其采用了如下方法进行数据整合。
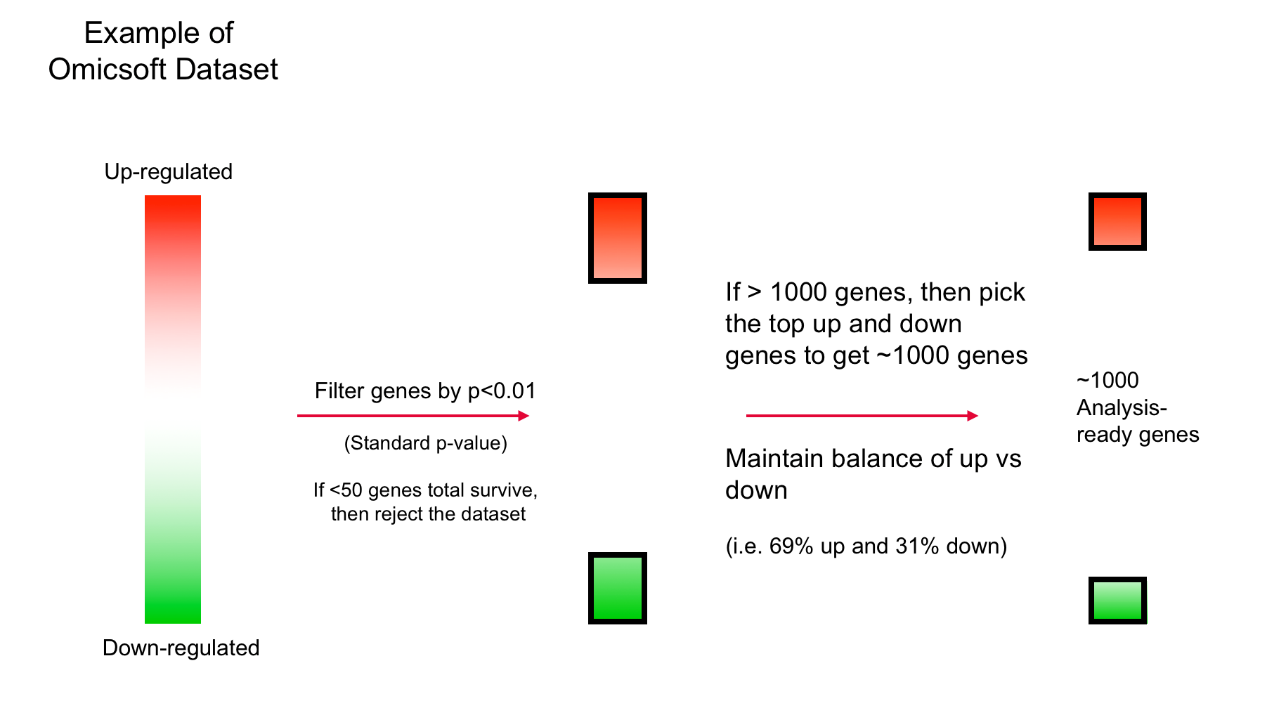
如何构建和比较数据集特征
在导入IPA数据库后,IPA对每个数据集进行Core analysis,并以如下策略构建特征集
Canonical Pathways (up to 20 pathways)
Upstream Regulators (up to 100 regulators)
Causal Networks (up to 100 master regulators)
Diseases & Functions (up to 100 diseases or functions)
具体构建策略如下图

需要注意的是,不是所有分析结果的每一个分析模块都有足够的特征值用于打分比较。比如某个分析结果只有6个Canonical pathway具有显著结果的z-score,那么其用于比较的通路特征值也就只有6个了。
分析数据集如何进行比较打分
和原先的计算打分类似,IPA在此依然采用类似的z-score进行实验数据集和后台背景数据集间的相似性打分:

通过以上计算过程的初步z-score并不会在结果中直接展示。为了将该数值变得更加可用,IPA会计算最大可能z-score来对结果进行标准化。

通过这个转换,最相似的数据匹配(也就是和自己匹配)的最大百分比不会超过80%,而较差的相似匹配可能就只有20%了。
通过这一系列计算步骤后,我们就可以非常轻松地在IPA中将自己的数据和49000多组其他疾病、癌症数据集进行比较、查询,了解其他类似研究都有怎样的新发现,为自己的研究提供思路、更重要的是还能让自己少走弯路、重复劳动。
![]() Red colored IgG4 caused by vitamin B12.pdf
Red colored IgG4 caused by vitamin B12.pdf
关注我们
服务热线: 18221381405
关注公众号
咨询微信客服

